2. 创建MultiAssayExperiment对象
本章节主要介绍如何将多组学项目数据导入分析流程,请根据您的数据情况按照以下导航进行:
2.1 创建多组学项目的MultiAssayExperiment对象
当用户处理多个组学项目的数据时,需要将数据导入并组合成一个MultiAssayExperiment对象。主要包含三个部分:①样本表型数据;②以SummarizedExperiment为格式的组学数据;③样本关系表。以下教程,将分别示例如何构建这三个部分。本教程也提供了部分示例数据用于学习。
示例数据:1. Github下载地址 2. 百度网盘下载地址
目录导航
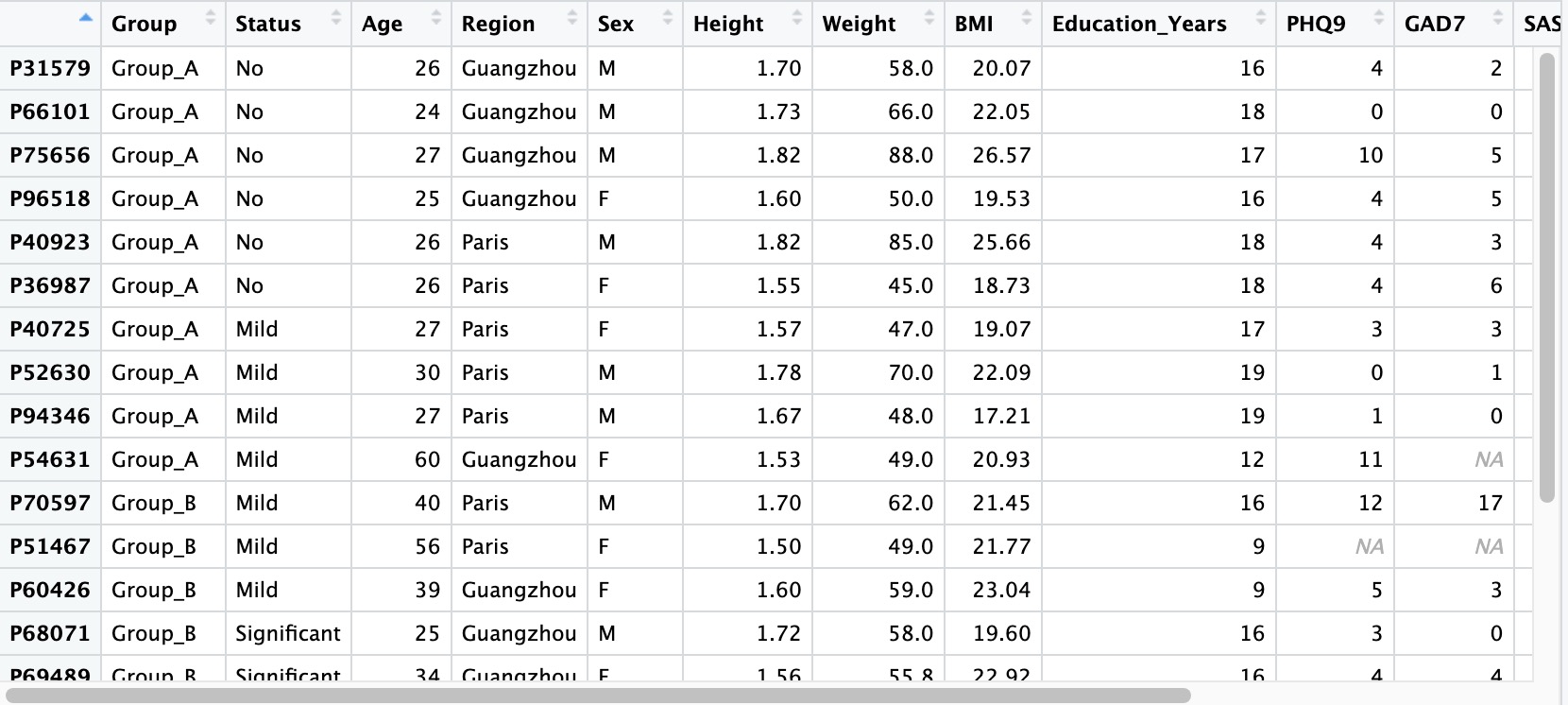
2.1.1 读取样本表型数据
样本表型数据是指受试者的基本信息,通常包括:受试者的一般人口学资料(性别、年龄、居住地等)、人体测量学资料(身高、体重、腰围等)、心理量表评分结果(焦虑量表、抑郁量表、睡眠量表等)、检验结果(血常规、肝功能、炎性因子等)等。
①样本相关数据中如果存在缺失值,请直接置空,不要用NA、-、missing等字符占位。
②样本相关数据的第一列请设置为样本编号,在读取时将其设置为行名(row.names = 1)。
③样本编号必须是唯一的,不能有重复。
④建议将数据中分类变量的取值设置为字符串,不要使用纯数字,避免引发不必要的问题。
⑤同一批受试者干预前后组学项目数据的编号方案,请参考10.3章节。
⑥样本名称请勿使用(-,^)等R不便于识别的符号,必要时使用下划线(_)替代。
## row.names=1 必须将样本名作为行名
meta_data <- read.table('col.txt',header = T,row.names = 1)
2.1.2 读取微生物组学数据
微生物组学数据是指通过各种微生物上游注释工具产出的、包含各个样本内微生物丰度信息的结果文件。常见的结果文件包括以下五种类型:ASV/OTU表格、界门纲目科属种级别丰度表、MetaPhlan和Humann产生的带有层级关系的丰度表、BIOM文件、QIIME2专属的QZV文件。模块EMP_taxonomy_import支持以上全部格式的结果文件输入,并将其转化为SummmariseExperiment对象。
2.1.2.1 ASV/OTU表格
此文件内第一行和第二行的#字符无需修改。数据文件内必须要包含taxonomy的物种注释信息。
①该数据文件必须包含名为
taxonomy列。②
taxonomy列必须包含微生物注释列。③建议使用
;对微生物注释进行分割。
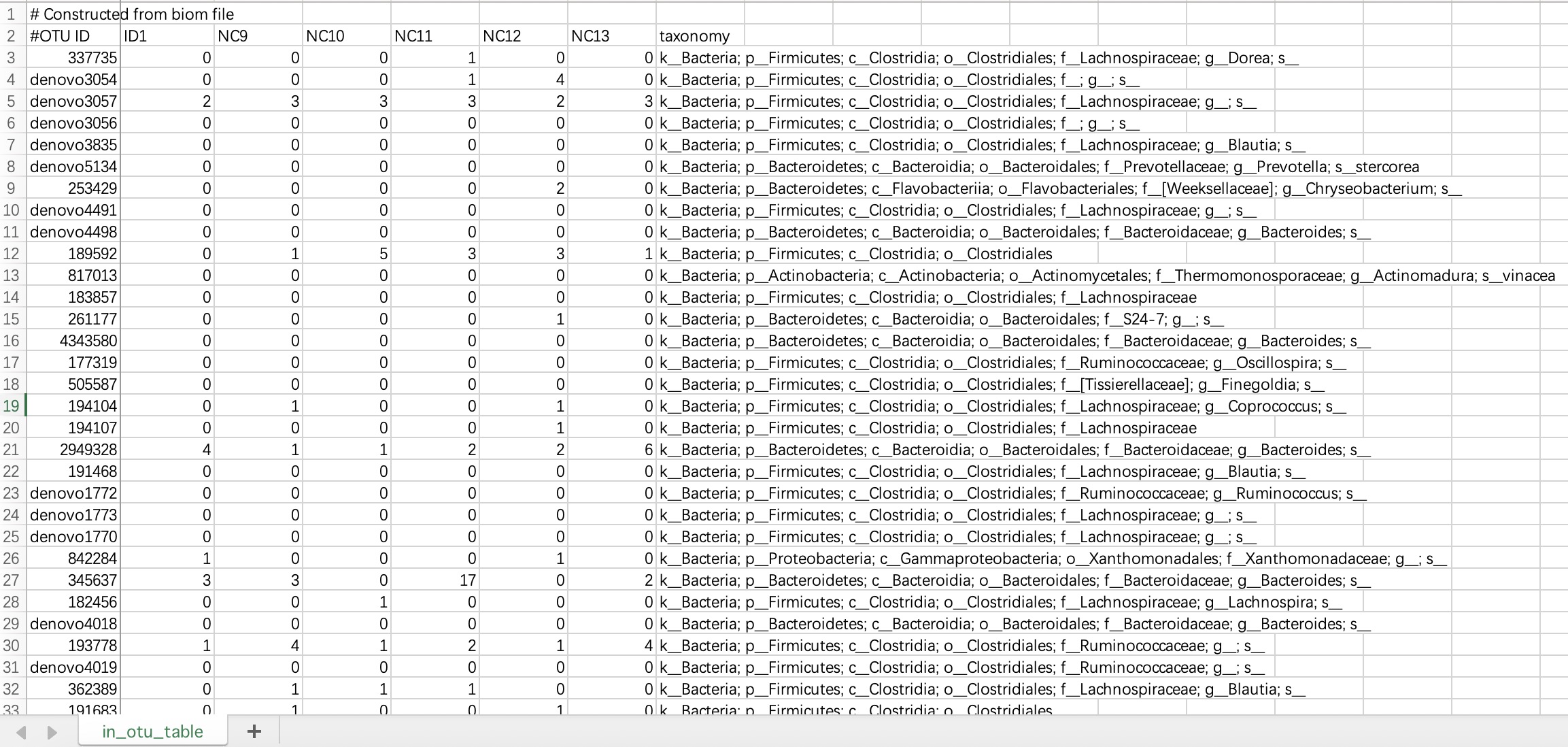
tax_data <- EMP_taxonomy_import('tax.txt',duplicate_feature=TRUE)
2.1.2.2 门纲目科属种级别丰度表
由于高分类级别数据可以转换为低分类级别数据(例如:Species数据可转换为Phylum数据),因此建议用户直接导入最高分类级别的数据。
模块
EMP_taxonomy_import的参数assay_name默认为counts(绝对丰度);如果实际数据为相对丰度,可输入relative(相对丰度)。
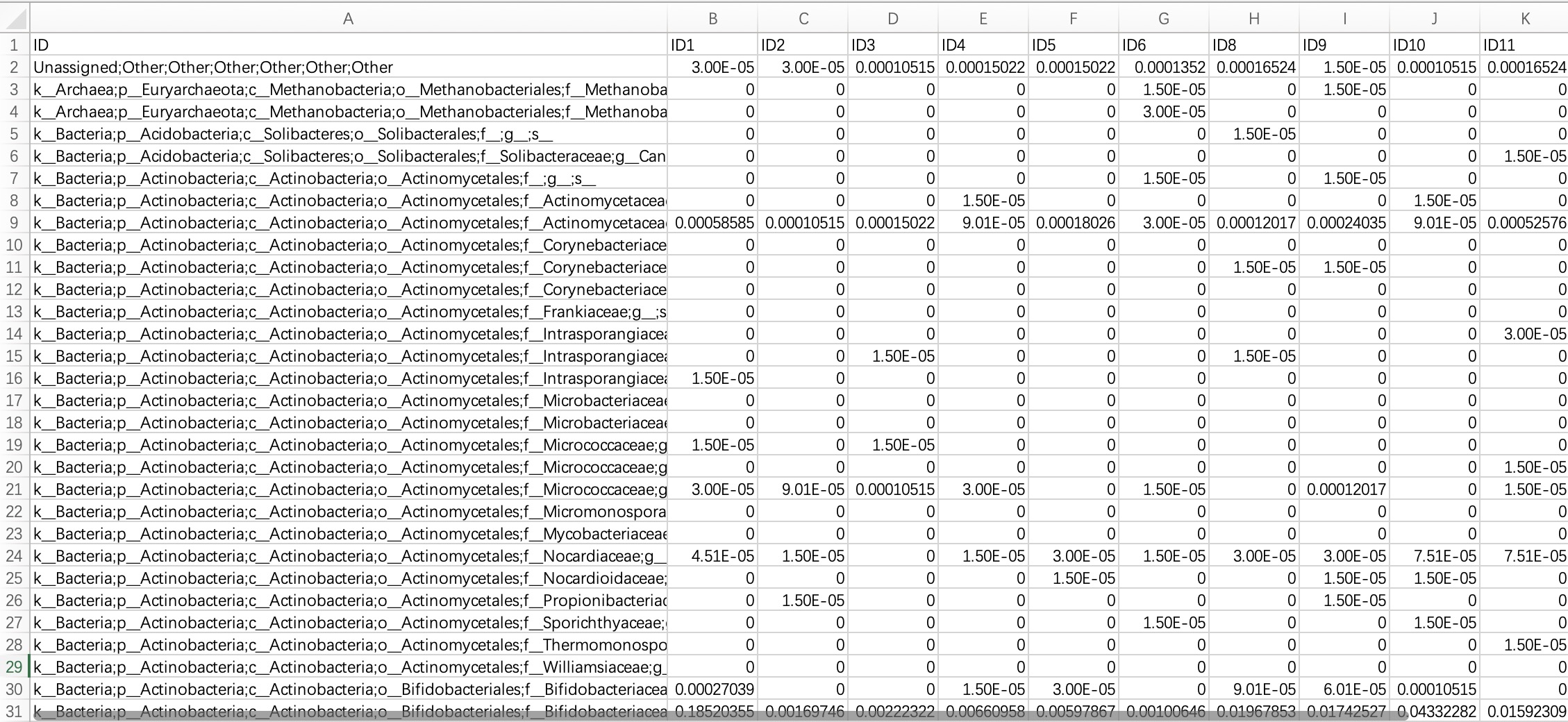
tax_data <- EMP_taxonomy_import('tax.txt')
2.1.2.3 MetaPhlan和Humann的分类注释表
此注释表的物种注释具有层级结构,通常包含界门纲目科属种各级别的全部结果信息。模块EMP_taxonomy_import可以自动识别最高分类级别的注释信息,并导入完整分类数据。
输入的数据为此种格式时,需要将在模块
EMP_taxonomy_import内指定参数humann_format=TRUE。
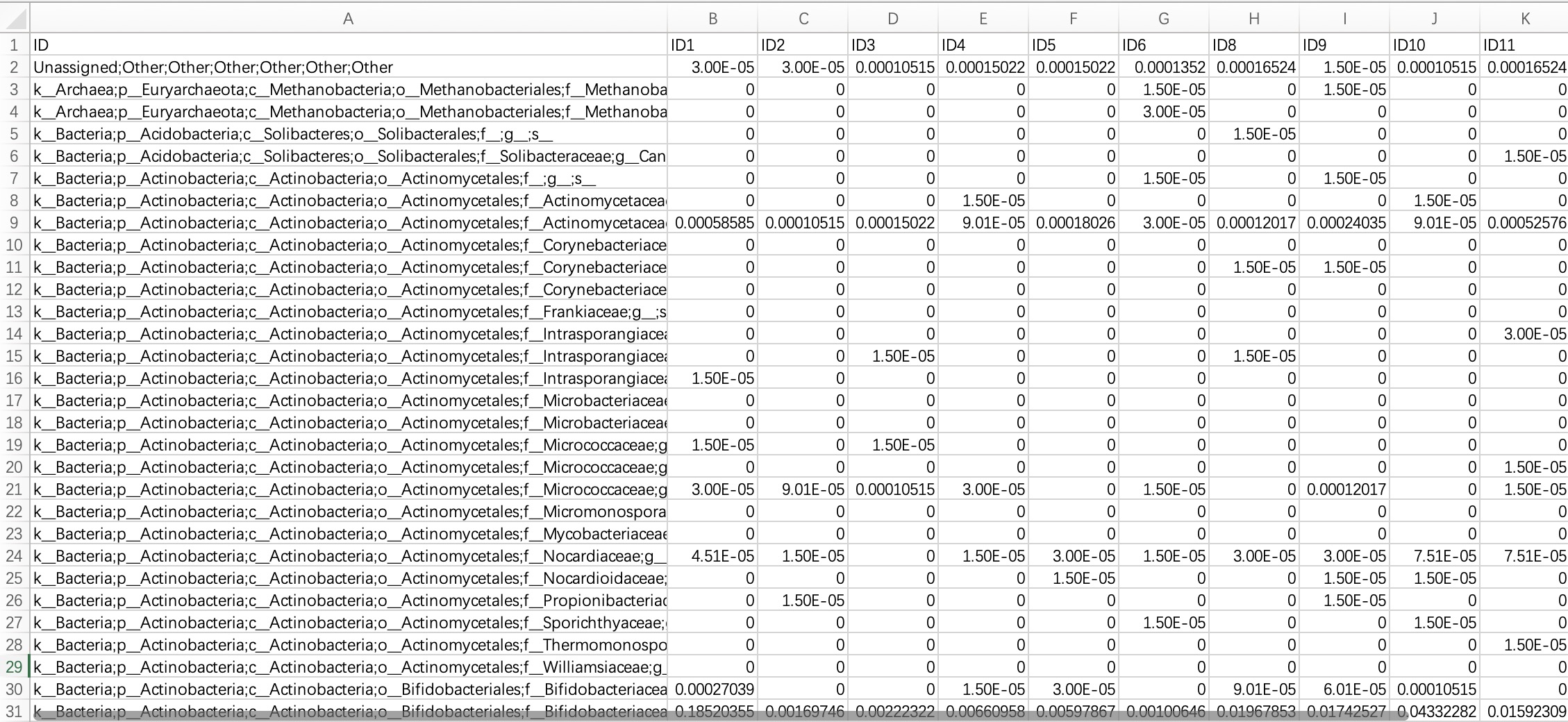
tax_data <- EMP_taxonomy_import('tax.txt',humann_format=TRUE)
2.1.2.4 Biom格式文件
Biom格式是QIIME1流程中常见的物种注释结果文件,部分用户也会将QIIME2流程的物种注释文件保存为biom格式便于数据存储。模块EMP_taxonomy_import可以直接读取biom格式文件,并导入数据信息。
输入的数据为此种格式时,需要将在模块
EMP_taxonomy_import内指定file_format='biom'。
tax_data <- EMP_taxonomy_import('tax.biom',
file_format='biom',duplicate_feature=TRUE)
2.1.2.5 Biom格式文件转化后的表格文件
Biom文件可以利用biom convert方法转换成表格文件,也可以直接导入。
输入的数据为此种格式时,无需修改表头和#符号。
tax_data <- EMP_taxonomy_import('tax.txt',duplicate_feature=TRUE)
2.1.2.6 qzv格式文件
在Qiime2流程qiime taxa barplot会产生微生物注释结果qzv文件。模块EMP_taxonomy_import可以直接读取qzv格式文件,并导入数据信息。
输入的数据为此种格式时,需要将在模块
EMP_taxonomy_import内指定file_format='qzv'。
tax_data <- EMP_taxonomy_import('tax.qzv',
file_format='qzv',duplicate_feature=TRUE)
2.1.3 读取KO/EC组学数据
KO/EC组学数据源于基于KEGG数据库的功能注释,主要包含KO(KEGG Orthology)和EC(Enzyme Commission)两种功能标识系统。此类数据通常由宏基因组测序结果经工具(如HUMAnN、PICRUSt2)分析所得,用于描述微生物群落潜在的代谢功能与通路活性。在导入时,工具支持读取标准的表格格式文件,其中行代表KO/EC功能特征,列代表样本,单元格中的数值通常为功能的丰度或计数。
主要注释类型:
KO (KEGG Orthology):KEGG直系同源群标识,用于关联基因功能与KEGG代谢通路。
EC (Enzyme Commission):酶学委员会编号,系统性地对酶进行归类与编码。
2.1.3.1 普通表格形式
普通格式的KO/EC组学项目数据是指宏基因组或宏转录组学注释的、以KO或EC编号注释的结果文件。模块EMP_function_import可以直接读取注释表格,并查询KEGG数据库整合对应的注释信息。
①编码中必须是纯KO和EC编号,例如:不能为KO:K00010,而必须仅为K00010;不能为ec:1.1.1.1,而必须仅为1.1.1.1。
②模块
EMP_function_import内参数assay_name默认为counts(绝对丰度),用户需要根据实际数据输入TPM、FPKM等。
ko_data <- EMP_function_import('ko.txt',type = 'ko')
ec_data <- EMP_function_import('ec.txt',type = 'ec')
2.1.3.2 Humann格式
Humann格式的KO/EC组学项目数据是指由Humann工具产生的分层KO/EC注释表。模块EMP_function_import会忽略UNGROUPED的信息,直接读取注释表格,并查询KEGG数据库整合对应的注释信息。
文件的表头需要清理干净,删除#等字符。
ko_data <- EMP_function_import('ko.txt',type = 'ko',humann_format = TRUE)
2.1.4 读取标准表格信息
标准表格数据通常是指转录组或代谢组等数据,这类数据没有特殊格式,可直接导入。
2.1.4.1 不含特征注释信息的数据导入
本数据常见于转录组或基因组数据,“行”为特征,“列”为样本。
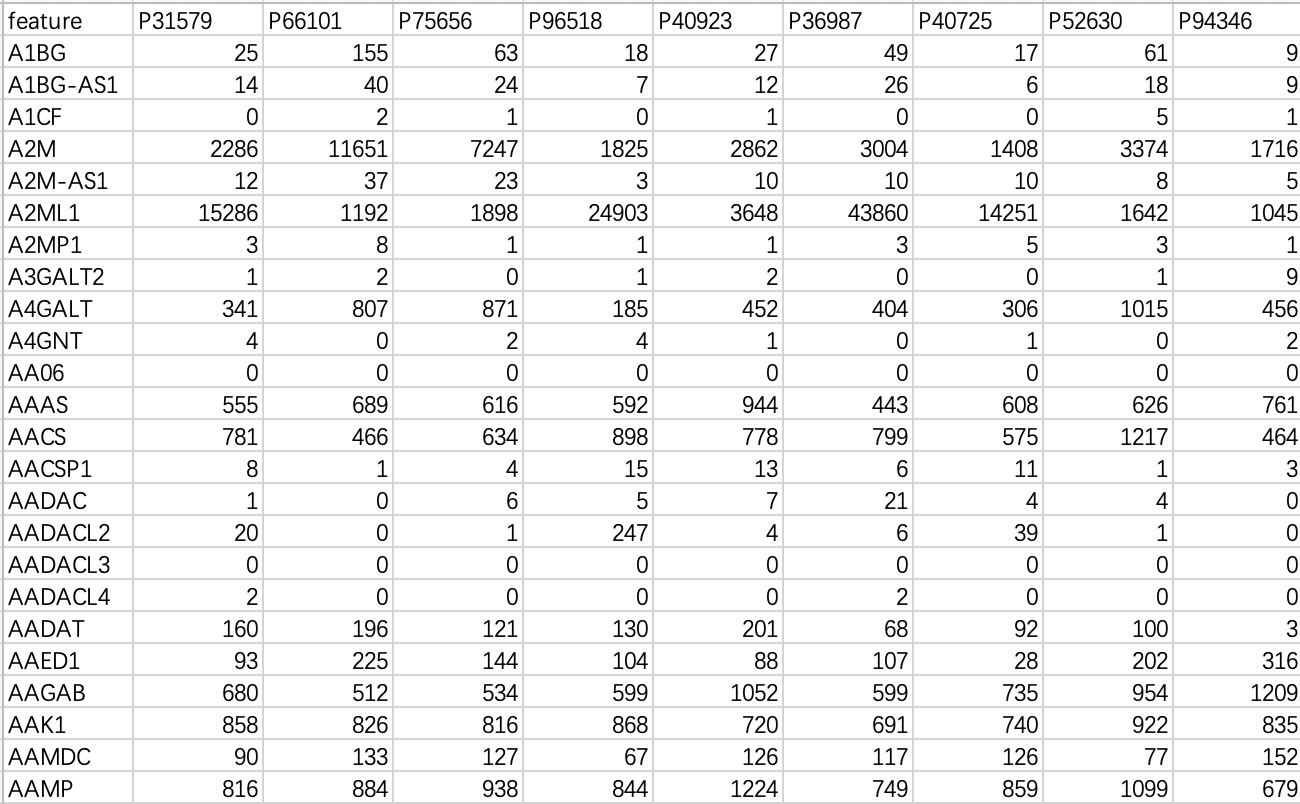
tran_data <- EMP_normal_import('tran.txt')
2.1.4.2 包含特征注释信息的数据导入
本数据常见于代谢组学数据,“行”为特征,“列”为样本编号和特征相关注释。导入这种数据时,需要指定样本编号列。
①可直接通过
sampleID指定样本编号。②可直接通过关系表指定样本编号。
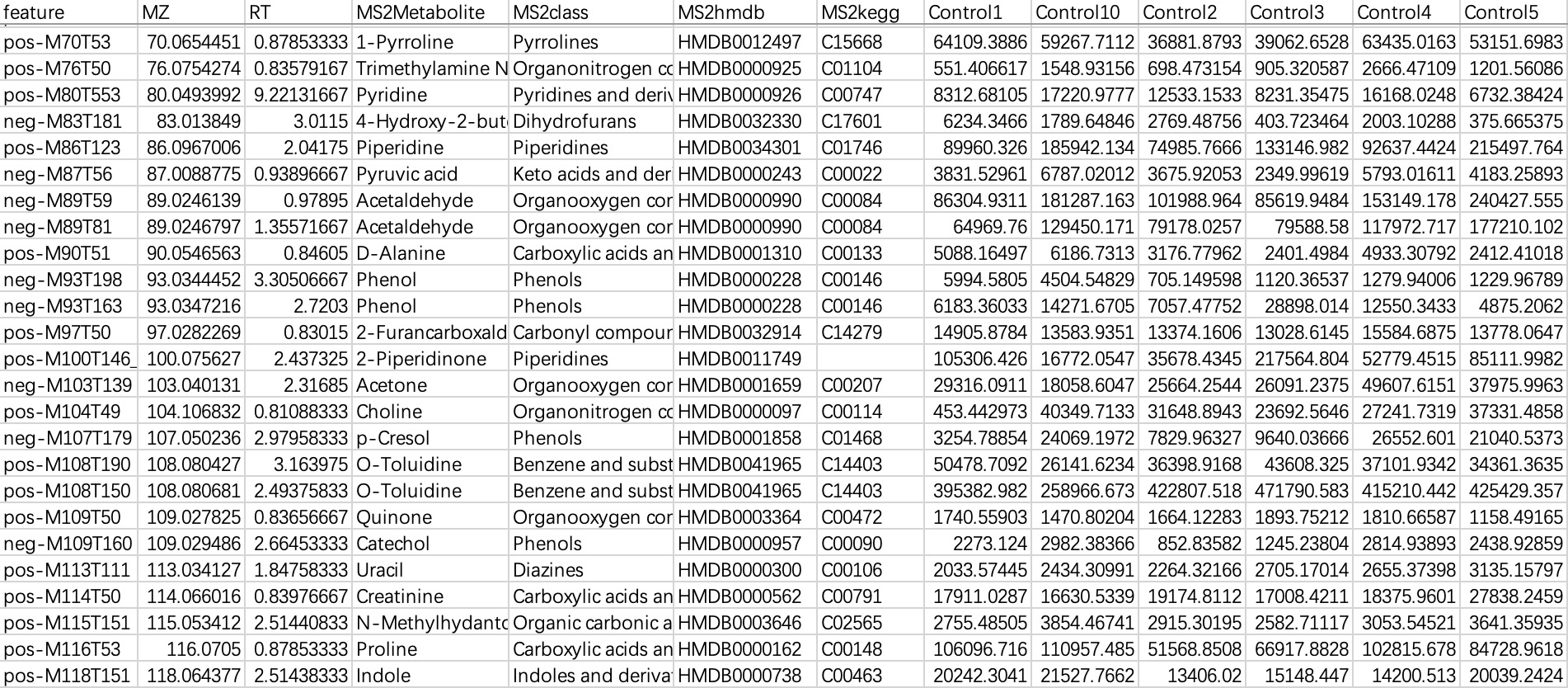
# 直接指定样本编号
sample_ID <-c("Control1", "Control10", "Control2", "Control3", "Control4",
"Control5", "Control6", "Control7", "Control8", "Control9",
"Treat10", "Treat1", "Treat2", "Treat3", "Treat4", "Treat5",
"Treat6", "Treat7", "Treat8", "Treat9")
metbol_data <- EMP_normal_import('metabol.txt',sampleID = sample_ID)
# 利用关系表来判断样本编号
metbol_data <- EMP_normal_import('metabol.txt',sampleID = sample_ID,
dfmap = dfmap,assay = 'untarget_metabol')
2.1.5 创建组学样本编号与受试者编号关系表
在完成数据导入后,我们已读取两部分关键数据:① 以SummarizedExperiment格式存储的组学数据,以及② 样本表型数据,接下来需构建的是③ 样本关系表。
该表的核心作用在于解决多组学整合中常见的样本命名不一致问题。在许多研究中,不同组学平台或实验批次可能对同一受试者使用不同的样本编号,导致表达矩阵与表型数据之间无法直接匹配。样本关系表正是为了建立两者之间的映射关系而设计。
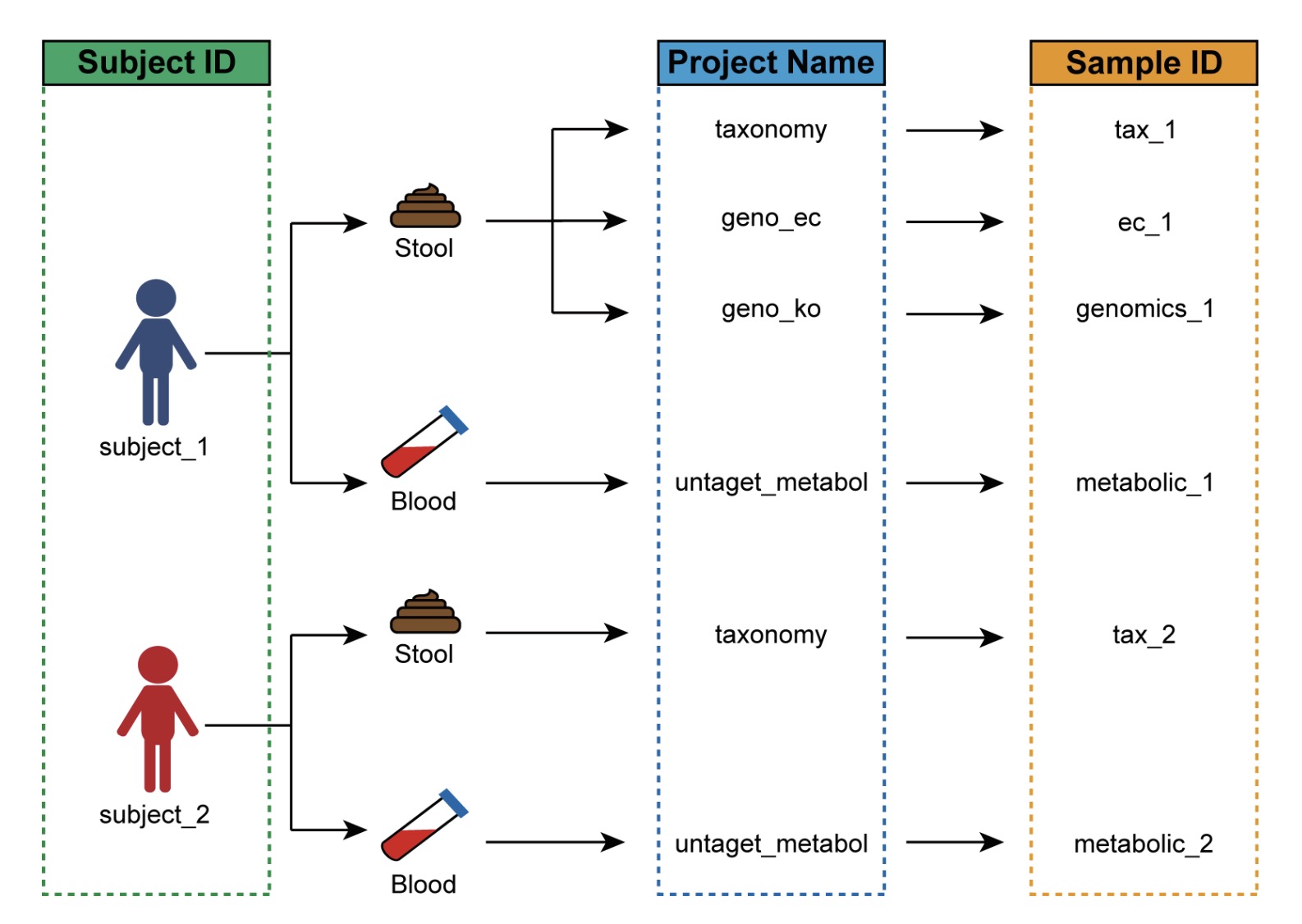
例如,在一项涉及多组学检测的人体研究中:
每位受试者有唯一的受试者编号(如P1、P2、P3…);
同一受试者可能采集多种生物样本,进行多种组学检测,如:
○ 16S rRNA物种注释(taxonomy),样本编号为 如tax1、tax2、tax3,…;
○ 微生物功能基因KO注释(geno_ko),样本编号为 genomics_1、genomics_2、genomics_3,…;
此时,必须通过样本关系表,将表型数据中的P1与不同组学数据中的tax_1 和genomics_1等样本编号进行关联。
一旦建立此表,用户即可将所有数据顺利整合至MultiAssayExperiment对象中。在后续分析中,EasyMultiProfiler包将依据该关系表,自动将各組学样本编号统一替换为受试者编号,确保数据整合的准确性与分析流程的顺畅性。
以关系表为基础的分析流程具有以下优势:
- 数据整合与一致性: 通过关系表,可以确保不同组学项目的数据集中的样本信息被正确地映射和整合,从而保证数据的一致性。
- 自动化处理: 可以自动化地管理和处理数据,无需手动修改,因此不用担心出现命名不一致的情况。
- 简化分析流程: 将数据整合并自动将样本编号替换为受试者编号后,分析流程更为流畅和清晰,减少了手动处理可能导致的错误。
如果组学表达矩阵中的样本名称与表型数据中的样本名称一致,则在关系表中创建同名即可。
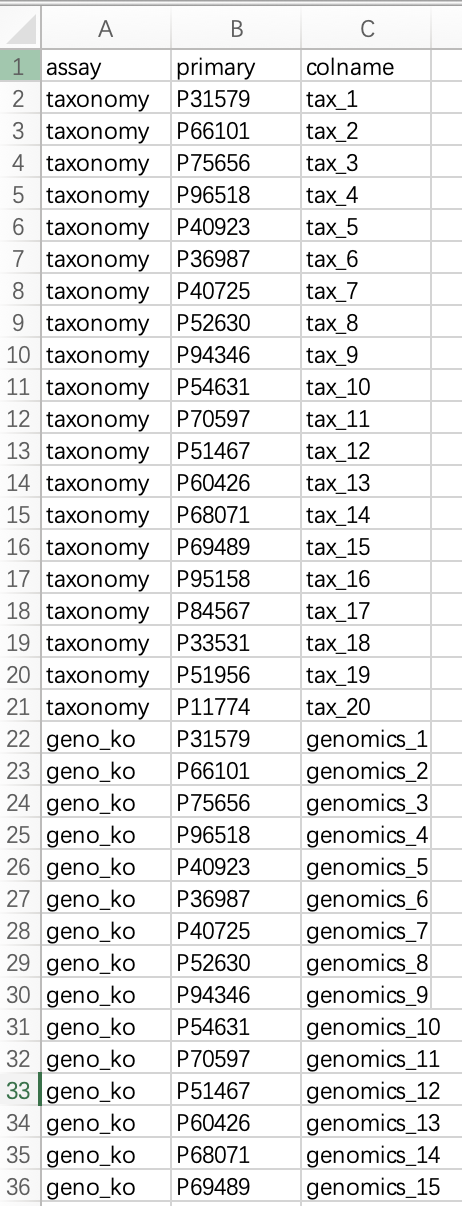
🏷️示例:创建受试者编号与组学项目样本编号的关系表。
①关系表必须由以下三列组成:assay(组学项目名称)、primary(受试者编号)、colname(组学项目样本编号)。
②assay(组学项目名称)指在这个数据集中你希望给组学的命名。primary(受试者编号)是表型数据中的样本名称。colname(组学项目样本编号)是所在组学的表达矩阵中样本的名称。
③每个组学项目的样本编号必须与关系表的colname逐一对应。
④同一批受试者干预前后组学项目数据的编号方案,请参考10.3章节。
此表需要保存为文件,读入R中。用户也可以手动在R内通过数据框直接创建。
dfmap <- read.table('dfmap.txt',header=TRUE,sep='\t')
2.1.6 整合全部数据导入MultiAssayExperiment对象
①本示例中的dfmap即2.1.5中的关系表。
②关系表内的
assay必须转换为因子。③
objlist内命名必须与关系表assay列的组学项目名称一致。④如组装失败,可以用模块
MultiAssayExperiment::prepMultiAssay(objlist,meta_data,dfmap)检查数据情况。⑤更多有关
MultiAssayExperiment包的详细信息可以参考此教程。
#### 关系表的assay必须转换为因子
dfmap$assay <- as.factor(dfmap$assay)
#### objlist内命名必须与关系表assay列的组学项目名称一致
objlist <- list("taxonomy" = tax_data,
"geno_ko" = ko_data,
"geno_ec" = ec_data,
"untarget_metabol" = metbol_data,
"host_gene" = geno_data)
MAE <- MultiAssayExperiment::MultiAssayExperiment(experiments=objlist,
colData=meta_data,
sampleMap=dfmap)
2.2 快速创建单组学项目的MultiAssayExperiment对象
当用户只有单一的组学项目数据(例如:微生物组学项目数据或转录组学项目数据),可通过模块EMP_easy_import快速构建MAE对象,直接进行下游分析流程。
①
type参数包含tax、ko、ec和normal四种方式,需要用户根据上文判断使用。② 由于单一组学项目不涉及创建关系表,因此表达矩阵的样本名称必须与表型数据中样本名称完全一致。
2.3 MultiAssayExperiment对象存放与读取
当创建完成MAE对象后,可以将其存入本地以便下次分析时直接读取,而无需重复创建步骤。
在存储前,请务必正确设置R的本地工作目录,数据文件将存储在该工作目录下。
# 保存在当地工作目录
saveRDS(MAE,file = 'MAE.rds')
# 加载已构建好的MAE容器
MAE <- readRDS(file = 'MAE.rds')
2.4 示例数据以及脚本
示例数据:1. Github下载地址 2. 百度网盘下载地址
library(EasyMultiProfiler)
## 加载表型数据
meta_data <- read.table('coldata.txt',header = T,row.names = 1)
## 加载样本表
dfmap <-read.table('dfmap.txt',header = T) %>%
dplyr::mutate(assay = factor(assay))
## 加载组学数据
tax_data <- EMP_taxonomy_import('tax.txt')
ko_data <- EMP_function_import('ko.txt',type = 'ko')
ec_data <- EMP_function_import('ec.txt',type = 'ec')
metbol_data <- EMP_normal_import('metabol.txt',dfmap = dfmap,assay = 'untarget_metabol')
geno_data <- EMP_normal_import('tran.txt',dfmap = dfmap,assay = 'host_gene')
## 组合成列表
objlist <- list("taxonomy" = tax_data,
"geno_ko" = ko_data,
"geno_ec" = ec_data,
"untarget_metabol" = metbol_data,
"host_gene" = geno_data
)
## 创建MAE对象
MAE <- MultiAssayExperiment::MultiAssayExperiment(objlist, meta_data, dfmap)
## 存储数据对象以备下次分析使用
saveRDS(object = MAE,file = 'demo_data.rds')
## 下次分析时直接读取对象,而无需再次组装
MAE <- readRDS('demo_data.rds')